
ESMO TAT Meeting
- 24 mrt 2022
- 6 minuten om te lezen
Bijgewerkt op: 25 mrt 2022
ESMO Targeted Anticancer Therapies Congress 2022
For many tumor types whole arsenals of therapies are now available, most strikingly for lung and breast cancer. Small molecule-based approaches target oncogenic drivers with increasing specificity either directly or indirectly via synthetic lethality. Oncogenic mutation-specific inhibitors and allosteric inhibitors as well approaches using targeted protein degradation are on the rise. Antibody-based approaches consist of immunotherapy and also come in advanced forms like ADCs and T-cell engagers. A third pillar is the use of cell therapies where the field is moving towards allogenic therapies that might eventually become scalable one-size fits all therapies that graft and persist well in patients. All these type of treatments seem to work even better when applied in the earlier treatment lines including (neo-) adjuvant settings. ADCs were leading the show with numerous ADCs producing profound clinical responses. Some experts believe that these type of agents can eventually replace chemotherapy, particularly when given early in the treatment line. The availability of many types of therapies in some of these tumors means that patients can be rapidly switched to different treatments in case of toxicity or drug-resistance. An urgently needed advance however is the universal clinical adoption of detailed and holistic analysis of the mechanisms of drug resistance for an individual patient. For instance, a lung cancer progressing on EGFRi can be the result of new EGFR mutations (for which other EGFRi may be available), or MET amplification (for which EGFR-MET bi-specifics or METi are available) or PI3K/RAS mutations (for which PI3K or KRAS inhibitors are available). Similarly, patients becoming resistant to ADCs due to surface antigen or payload target mutation can be switched to different ADCs. The mindset of many drug developers is still to move new drugs into Phase II using a dose that is near the MTD, a strategy dating back from the chemotherapy era. However, these novel treatment paradigms have dose-response and dose-toxicity relationships that can be very different from chemotherapy. For instance target saturation may occur well below the MTD or increase unwanted on-target toxicities at high doses (e.g. CLTA4 blockade at Tregs).
Impressive clinical data of multiple new small molecule therapies were reported. The field is moving away from dirty multi-kinase inhibitors to kinase family specific (e.g. FGFR, TRK), isoform-selective (e.g. MET) or even mutation-specific inhibitors (KRASG12C, PDGFR842V). Mutation-specific inhibitors invoke resistance more quickly whereas highly potent family kinase inhibitors or isoform-specific inhibitors may be more durable but can induce on-target toxicity (e.g. MET, ROS). Kinase inhibitors can be rationally combined with other therapies based on complementary MoAs whilst at the same time avoiding overlapping toxicities. For instance, good results are obtained in colorectal cancer when KRASG12Ci are combined with EGFRi given that the EGFR pathway is a strong resistance mechanism to KRASG12Ci in colon cancer specifically.

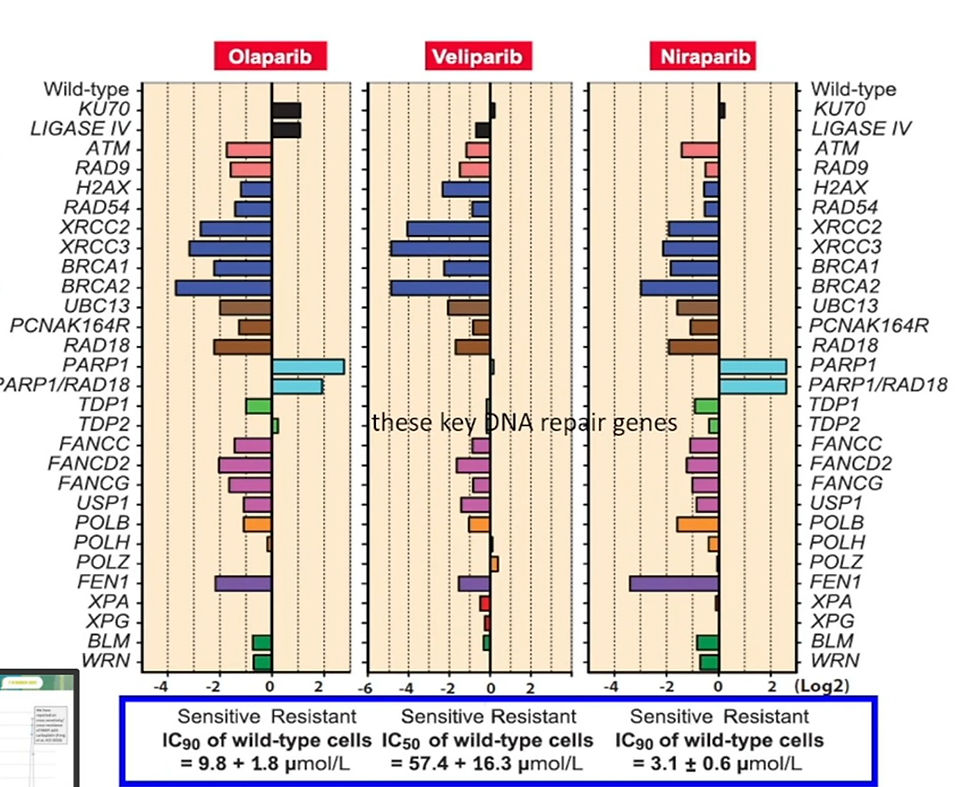
Novel approaches are being developed to target the other KRAS mutations by molecular glue (MG) and PROTAC-mediated protein degradation. The most advanced protein degrading small molecules are PROTACs targeting androgen (AR) and estrogen receptor (ER). Clinical data is so far anecdotical however, albeit it is encouraging to see that these agents are active against mutant AR and ER. Novel PROTACs degrade targets like SMARCA2/4 or even come in trivalent forms. The field of synthetic lethality is still much focused on vulnerabilities created by loss of DNA repair enzymes like BRCA1/2. It is becoming apparent that the activity of PARPi is not just confined to tumors that have lost homologous recombination repair (HRR) enzymes including BRCA1/2 and PALPB2 but also to tumors that have lost enzymes active in other forms of DNA repair (e.g. ATM and RNASEH2B) or chromatin cohesion. For example, tumors that have lost XRCC3 are even more sensitive to olaparib than tumors that have lost BRCA1/2. In all these case, homozygous loss of the respective (so not mutation) appears to lead more durable responses to PARPi. The most studied combination therapy is PARPi and ATMi that synergize in tumors deficient in ATM, BRCA1/2 but also p53/Rb. Overlapping toxicities limit the use of PARPi and ATMi but new dosing and scheduling strategies as well as use of novel biomarkers of sensitivity may circumvent this problem.
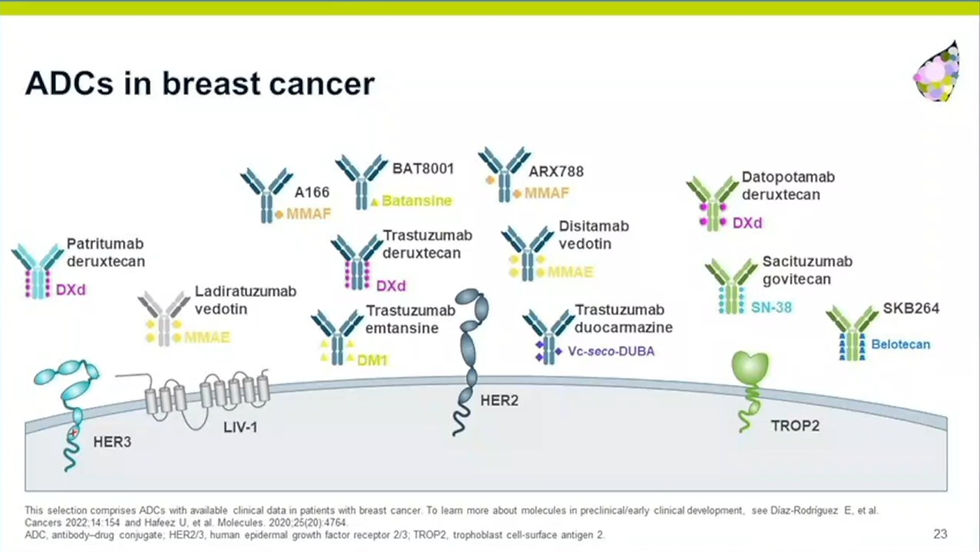
Many examples of profound clinical responses of ADCs were demonstrated in lung cancer and breast cancer in particular. Since multiple ADCs targeting different target antigens and using different payloads are on the market for these cancers, the field is moving towards combining ADCs or switching patients to a different ADC once patients develop resistance. Patients can become resistant to ADCs by mutations in the receptor, endocytosis pathway or target of the payload. ADCs are also being rationally combined with molecularly targeted agents that synergize on the same biological pathway, for instance ADCs with topoisomerase inhibitors in combination with PARPi that act synergistically in DNA damage induction. These type of combinations would not be possible in unconjugated forms given the overlapping toxicities. It is clear that particularly those ADC payloads that can create a bystander effect on adjacent tumor and vasculature are clinically most active. Early impressive clinical data in bladder and breast cancer suggests that when such payloads are also immunogenic, they form attractive combination partners with anti-PD1. Another advantage of payloads that can create bystander effects (either by having a labile linker or by being able to diffuse across membranes) is that they are also active in tumor cells with low antigen expression. An important realization is that mid-high potency payloads like deruxtecan allow a much better therapeutic window for targets that are no so tumor-selective. In terms of ADC design constraints, the field is moving towards site-specific payload conjugation (i.e. leading to improved stability), novel types of warheads (BCL2i, radio-pharmaceuticals, i.e.) informed by a deep understanding of uptake-internalization-target engagement of novel drug-linkers.
Bi-specific T-cell engagers readily create bystander effects since these agents recruit T-cells to the tumor micro-environment. A limitation is that T-cell engagers need a more tumor restricted antigen expression to enable an acceptable therapeutic window and also are dependent on the availability of T-cells in the TME. Thus, T-cell engagers seem not suited for cold tumors or for targeting antigens of limited specificity (e.g. HER2). However, trivalent engagers are being developed that use “dirty” antigens like HER2 in combination with a tumor-selective antigen. Another obvious development is adding checkpoint inhibitor functionality to T-cell engagers, either in combo with anti-PD1 (more control on toxicity) or building it in the T-cell engager product (single product but complex to engineer, potentially better activity in the TME). An exciting new field are TCR mimetic antibodies that recognize cancer cells expressing intracellular antigens via MHC molecules. Current antibody-based approaches only target surface antigens, but most tumor-specific antigens are actually expressed intracellularly. TCR-like antibodies can trigger ADCC, CDC, antibody-dependent cellular phagocytosis (ADCP), or the direct induction of apoptosis. Immunocore’s Kimmtrak (tebentafusp-tabn) is the first of such therapies which is approved for unresectable or metastatic uveal melanoma.

The field of allogenic cell therapy has been making progress. Strategies include genetic deletion of TCR, HLA genes and other factors that mediate graft versus host disease (GvhD) that is a key limitation of T-cell based allogeneic cell therapy. First clinical PoC studies showing durable remissions with genome-edited allogenic T-cell therapy were presented. Unlike T-cells, NK cells don’t induce GvhD as they don’t have TCRs and HLAs. Furthermore, NK cells are not dependent on HLA-expressed antigens for killing cancer cells. Also here, promising clinical PoC studies with CD28/41BB enhanced NK cells were presented. Crispr/Cas9-mediated gene editing however induce low-frequency chromosomal translocations which may result in cancerous cell therapy products. The technology is being optimized and other gene editing techniques are also being developed. As I reported earlier, hurdles that need to overcome include: increasing adoption and persistence of the cell therapy in the host (particularly a problem for NK cells), tumor escape by antigen editing (e.g. loss of CD19) and immune-suppressive signals in the TME (solid tumors). In terms of other I-O approaches, no new innovations were reported beyond the new emerging T-cell checkpoint inhibitors (e.g. TIGIT, LAG3) that were discussed at the ESMO I-O meeting in 2021. Agents targeting metabolism (adenosine, kynurine) so far don’t confer single-agent activity in the clinic. Potentially the profound metabolic plasticity in TMEs may limit their usefulness. Some early data was shown of small molecules targeting enzymes involved in T-cell signaling.

In conclusions, oncologists have increasingly more treatments to them available. The selection of the right treatment should be based on certain TME parameters of the tumor (cold/hot, T-cell infiltrated, PD1 expression etc), the availability of actionable genetic mutations, cell surface antigens and biological markers (e.g. integrity of DNA damage repair mechanisms). These type of evaluations should be done as soon as the patient is diagnosed with cancer but also during the course of treatment evolution when drug resistance occurs.
Opmerkingen